
Introducing Reaction Condition Prediction
5 minute read
As mentioned in my bio, my PhD focuses on a project titled: ‘Prediction of optimal reaction conditions using Artificial Intelligence tools’. But what exactly are ‘optimal reaction conditions’?
Reaction conditions are the environmental factors under which a chemical reaction proceeds. For those unfamiliar with chemistry, a chemical reaction is a process where one or more substances (reactants) are converted into one or more different substances (products).
Why are chemical reaction conditions important?
The conditions of a chemical reaction are crucial for several reasons. In most cases, we can’t simply mix the building blocks of our desired product and expect it to form. Specific reaction conditions are required to:
- Initiate the reaction
- Ensure correct regioselectivity (reaction occurs at the intended location on the molecule)
- Achieve proper stereoselectivity (resulting in the correct 3D structure, which is particularly important in drug synthesis)
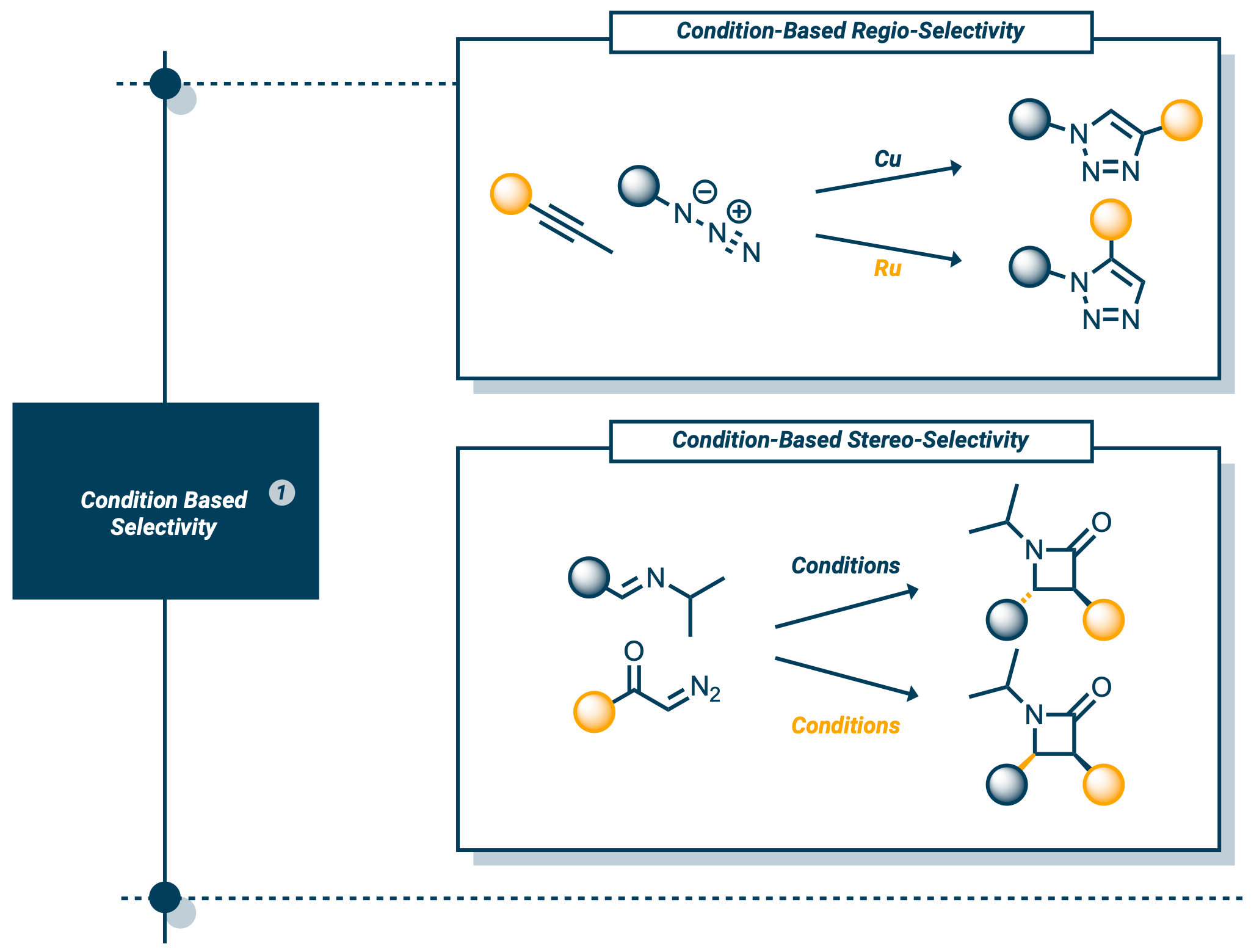 |
|---|
| Examples of how different conditions lead to different products. Top: Different conditions result in varying positions of R groups, potentially affecting the molecule’s properties. Bottom: Bond colors indicate stereochemistry (3D position). Different conditions yield different stereochemistries, which can significantly impact properties - the infamous case of Thalidomide illustrates this point. |
Beyond producing the desired product, reaction conditions also influence:
- Yield: The amount of product produced, which is crucial for economic viability.
- Cost and environmental impact: Considerations include the use of expensive catalysts or environmentally harmful solvents.
- Synthesis continuity: In multi-step syntheses, particularly for pharmaceuticals, insufficient yield at one stage can halt the entire process, wasting resources.
Optimising reaction conditions is therefore a balancing act between selectivity, yield, cost-effectiveness, and environmental considerations.
What approaches exist already?
Let’s explore various approaches to selecting reaction conditions, including both traditional methods and those leveraging machine learning.
Experimental Approaches
Traditionally, synthetic chemists rely on their expertise to perform an ‘intuitive’ similarity search. This involves scouring literature for similar reactions and using those conditions as a starting point. But what happens when these conditions don’t work?
One common method is ‘One Factor At a Time’ (OFAT). This involves systematically altering one dimension of the reaction conditions to optimise yield. Once an optimal point is found for one factor, it’s fixed, and another condition is varied. This process continues until all conditions have been ‘optimised’.
However, OFAT has significant limitations:
- It ignores the interdependence between different reaction conditions.
- It’s often inaccurate and inefficient.
- It can miss the global optimum for a set of conditions.
 |
|---|
| Illustration of OFAT’s limitations. The method fails to capture how yield changes due to the interdependence of different conditions. Dotted lines represent varied conditions, with experiments at each dot. The redder the line in the contour plot, the higher the yield. |
More sophisticated approaches like ‘Design of Experiments’ (DoE) can explore reaction space more efficiently than OFAT. However, computational approaches offer the potential to significantly reduce time and costs by screening conditions before lab work begins.
Computational Approaches
We can categorise computational methods into five main groups. All of these models will follow a similar workflow, which can be seen below:
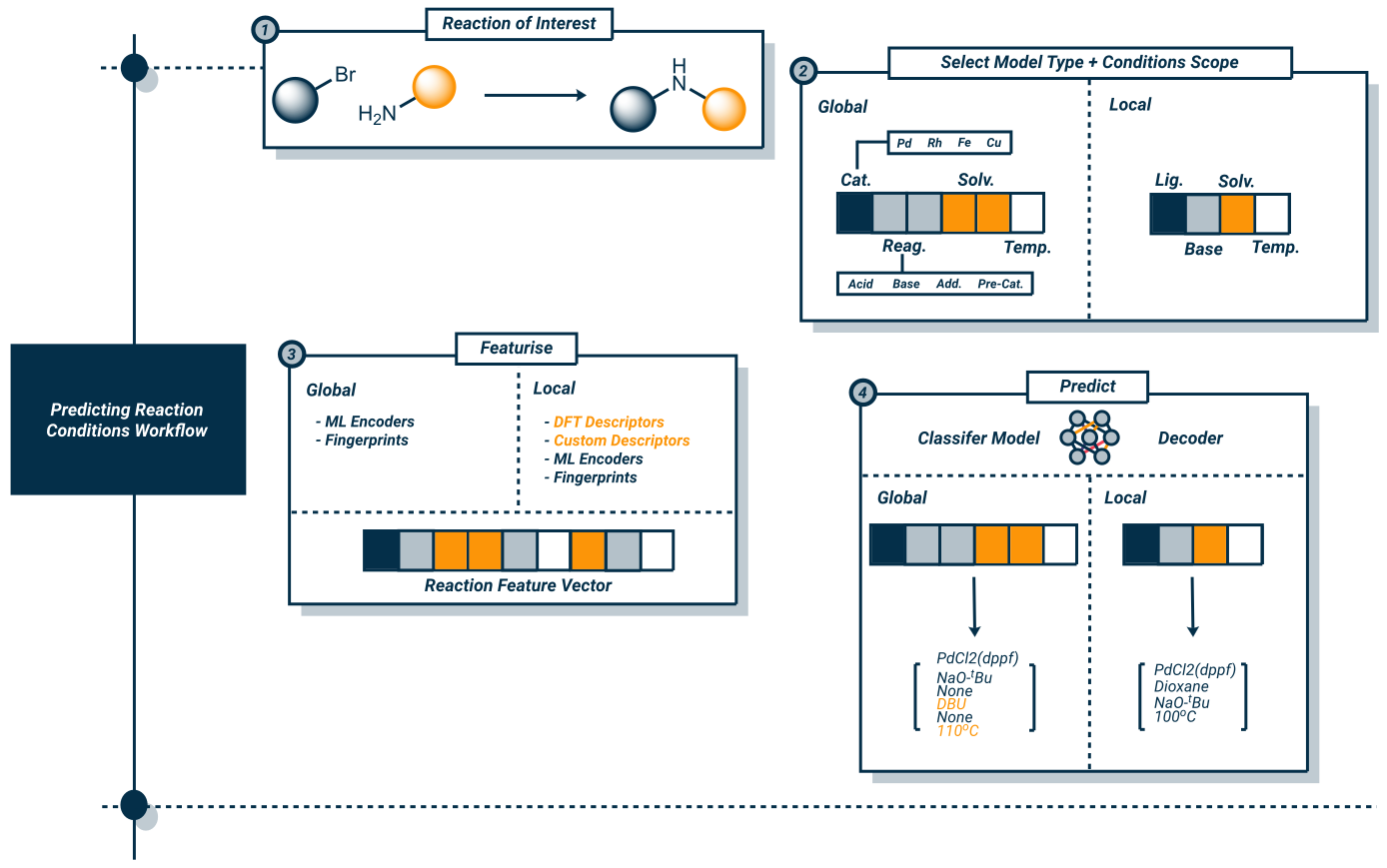 |
|---|
| An example modelling workflow for predicting reaction conditions, in this figure ‘Global’ and ‘Local’ data/models refers to the scope of the data/models, which we will discuss in a later blog. |
Similarity-Based Models
These models enhance the ‘intuitive’ similarity search by leveraging large databases of chemical reactions. They provide simple, interpretable suggestions for new substrates. However, they have limitations:
- Slow performance on large databases
- Inability to generalise to new combinations of existing conditions
Ranking Models
Ranking models work differently:
- The user inputs a reaction.
- The model defines a search space of possible conditions.
- All possible combinations of reagents are enumerated into condition sets.
- The model ranks these conditions and returns the best options.
Advantages:
- Can generalise outside of known condition combinations
Disadvantages:
- Computational limitations when dealing with many reaction classes due to the explosion of possible combinations
Classification Models
These models treat condition prediction as a multi-class classification problem:
- Conditions are encoded as one-hot vectors
- Can cover multiple reaction classes effectively
- Don’t require enumeration of all possible conditions
While some chemical information is lost in the encoding, these models have shown strong performance across various datasets.
Transformer-Based Models
Leveraging the power of attention mechanisms, these models:
- Encode reactions as SMILES strings (text-based molecular representations)
- Treat the problem as a sequence-to-sequence translation task
- Show state-of-the-art accuracy on large datasets
However, they require substantial amounts of data, making them less suitable for novel reaction types with limited data.
Yield-Based Models
Yield prediction is a closely related field to condition prediction. With accurate yield prediction tools, we can:
- Enumerate possible conditions
- Predict the yield for each set
- Select the highest-yielding conditions for testing
This approach is often used in conjunction with High Throughput Experimentation (HTE) in an ‘Active Learning’ workflow, where experimental results continually update and improve the models.
Conclusion
Reaction condition prediction is a complex, multifaceted optimisation problem. The interdependence of various factors means that changing one condition can significantly alter the impact of others. As we continue to develop more sophisticated computational tools, we’re moving closer to being able to reliably predict optimal conditions for a wide range of chemical reactions, potentially revolutionising the field of synthetic chemistry.